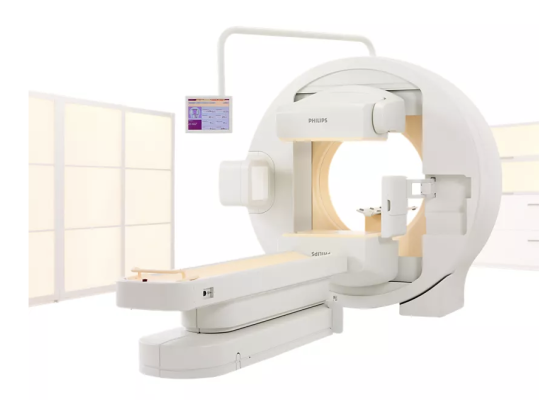
Image courtesy of Philips
February 15, 2024 — According to a release issued by the U.S. Food and Drug Administration (FDA), Philips is recalling its BrightView, BrightView X, and BrightView XCT, as the detector may unexpectedly fall due to a component failure. Update as of 12:59 pm 2/15/24 from the FDA: Please be aware, this recall is a correction, not a product removal.
Use of the affected device may cause a person to experience neck injury, contusion, traumatic brain injury/concussion, death, crush injury, fracture, laceration, muscle or ligament sprain/strain, as well as an interruption to the BrightView system operation. If detector is positioned above the large opening of the device (the center of the gantry), there may be an interruption to normal system operation.
The FDA has identified this as a Class I recall, the most serious type of recall. Use of these devices may cause serious injuries or death. Please be aware, this recall is a correction, not a product removal.
Recalled Product
- Product Names: BrightView, BrightView X, BrightView XCT
- Product Codes: See Recall Database Entry
- Model Numbers:
- BrightView: 882480; 453560279781, 453560279791, 453560279811, 453560279801, 2170-3000A, 2170-3001A, 2170-3002A, 2170-3003A
- BrightView X: 882478; 453560824741, 453560829261
- BrightView XCT: 882482; 453560462131, 453560749161
- Manufacturing Dates: September 2007 to June 2013
- Distribution Dates: November 29, 2007 to June 5, 2013
- Devices Recalled in the U.S.: 553
- Date Initiated by Firm: December 15, 2023
Device Use
The Philips Brightview system is a Single Photon Emission Computed Tomography (SPECT) machine used to take images showing biological activity in the human body, for medical personnel to review. The BrightView XCT model combines SPECT and Computed Tomography (CT) imaging.
Reason for Recall
Philips is recalling their BrightView, BrightView X, and BrightView XCT, as the detector may unexpectedly fall due to a component failure.
A falling detector may cause a person to experience neck injury, contusion, traumatic brain injury/concussion, death, crush injury, fracture, laceration, muscle or ligament sprain/strain, as well as an interruption to the BrightView system operation. If detector is positioned above the large opening of the device (center of the gantry), there may be an interruption to normal system operation.
There has been one reported incident from use of this device, and no reports of injuries or deaths.
Who May be Affected
- People who receive scans using a Philips BrightView, BrightView X, BrightView XCT.
- Health care providers who use BrightView Camera Systems to obtain detailed pictures or measurements of the human body.
- Field service engineers who service a Philips BrightView, BrightView X, BrightView XCT.
What to Do
On December 16, 2023, Philips sent all affected customers an Urgent Medical Device Correction letter.
The letter requested all affected users to not position a patient's lower limbs directly under the detector, below the center of the gantry. If the detector is above the center of the gantry and the detector support component fails, the detector will not be able to move to complete the imaging.
The letter also described two scenarios:
- Scenario 1: Detector positioned below the center of the gantry: If the patient’s lower limb(s) is directly below the lower detector and the support component fails, the detector may descend downward in an uncontrolled manner and contact the patient.
- Scenario 2: Detector above the center of the gantry: If the support component fails, the detector will remain in place, and will not move as intended for clinical imaging, resulting in an interruption to normal system operation. A rescan or re-injection of radiopharmaceutical to the patient may be required.
Philips will contact customers to schedule a time for a Philips Field Service Engineer (FSE) to visit the customer site and correct the system if necessary.
Contact Information
Customers in the U.S. with questions about this recall should contact the Customer Care Solutions Center at 1-800-722-9377.
Additional Resources
- Medical Device Recall Database Entries
How do I report a problem?
Health care professionals and consumers may report adverse reactions or quality problems they experienced using these devices to MedWatch: The FDA Safety Information and Adverse Event Reporting Program using an online form, regular mail, or FAX.
 August 06, 2024
August 06, 2024