
Greg Freiherr has reported on developments in radiology since 1983. He runs the consulting service, The Freiherr Group.
Blog | Information Technology | December 17, 2015
Will the FDA Be Too Much for Intelligent Machines?

Machine learning is already more our lives than many people realize. Smart algorithms complete phrases and correct misspellings typed into Google’s search engine; recognize faces in photos on Facebook; suggest movies on Netflix. Now algorithms are being groomed to help radiologists find disease in patient images and optimize scans.
Deep learning algorithms show promise for every type of digital imaging. A San Francisco-startup called Enlitic has already deployed engineers to implement one such algorithm in Australian and Asian imaging clinics. Meanwhile, IBM is grooming Watson Health to help physicians make diagnoses. Intelligent machines may one day take the reins during the exam itself, optimizing protocols on the fly to home in on pathology.
While this all sounds like artificial intelligence is right around the corner, it actually isn’t. Not because software programmers don’t have the skills. Or there is some intractable technological. It’s because the U.S. Food and Drug Administration (FDA) won’t let it, at least not any time soon.
For a glimpse of the minefield that awaits deep learning algorithms, look no further than the history of computer aided detection in digital mammography.
In 2012, the FDA released two guidance documents for companies and its own staff regarding computer-assisted detection devices used in radiology. The first recommends the documentation and performance testing for “CADe” devices cleared under the agency’s 510(k) process; the second recommends criteria for clinical performance studies for CADe devices cleared under 510(k) rules, as well as "CADx" devices approved under the premarket approval (PMA) process.
To help keep them straight, here’s how the FDA defines these two types of devices. CADe is defined as “computerized systems that incorporate pattern recognition and data analysis capabilities (i.e., combine values, measurements or features extracted from the patient radiological data) and are intended to identify, mark, highlight or in any other manner direct attention to portions of an image, or aspects of radiology device data, that may reveal abnormalities during interpretation of patient radiology images or patient radiology device data by the intended user.”
CADx is defined by the FDA as “computerized systems intended to provide information beyond identifying, marking, highlighting or in any other manner directing attention to portions of an image, or aspects of radiology device data, that may reveal abnormalities during interpretation of patient radiology images or patient radiology device data by the clinician.”
Three years passed after the FDA published draft guidance for these devices. This draft was preceded by years more during which industry plodded through the bureaucratic quagmire on Fishers Lane in Rockville, Md., asking the agency to come up with formal guidance that might give them some rules to follow when submitting marketing applications for new CAD devices.
Computer-aided detection algorithms for digital mammography spurred this action. Remarkably, the first guidance released by the FDA in 2012, the one focused just on the clearance process, does not even mention mammography CAD. The second, which addresses the performance of clinical studies for clearance and approval, does — but as a type of CADx, if the mammography software is “designed both to identify and prompt potential microcalcification clusters and masses on digital mammograms, and to provide a probability of malignancy score to the clinician for each potential lesion as additional information.”
With no clear alternative, companies making mammography CAD products have continued to submit applications to the FDA for review under the premarket approval process, the most rigorous process that the FDA demands of medical device manufacturers. Arguably, the FDA has softened the blow for companies whose initial devices have been premarket approved. Iterative improvements require only a “PMA supplement.”
But for first timers with a mammography CAD, the ins and outs of this regulatory process can be daunting. And that is what the developers of AI products will face … in bold letters.
While it’s not exactly known what deep learning algorithms will do — or how — they will do it, they definitely will go beyond the capabilities of the current generation of CAD algorithms. And that is the point.
Despite years of trying, industry was unable to get the FDA to identify mammography CAD as a CADe, which would formally allow these algorithms to be reviewed under the 510(k) clearance process. The chances are virtually zero that companies developing deep learning algorithms will succeed in getting the FDA to classify them so they can be reviewed under the 510(k) process — if not out of principle then by definition of the 510(k) process itself.
Why? Because DL programs will be submitted for FDA review as the first of their kind. They will lack the “predicate” devices needed for them to be considered under the 510(k) system, whose cornerstone is to gauge equivalency to devices already on the U.S. market.
This means — without a shadow of a doubt — that DL products will have to be reviewed under the FDA’s premarket approval process. And that is going to be tough. The FDA is not going to like that these learning algorithms are being designed to write their own rules for spotting disease and then to rewrite them to fit new experiences. Even for the simplest CAD products the FDA recommends “you provide information on the algorithm design and function including details on algorithm implementation” and “briefly describe the design and function for each stage of your algorithm.”
Given this, the developers of DL algorithms have to start working with the FDA now, if they want their products on the U.S. market in the foreseeable future. They will have to figure out what the FDA wants to see in their applications and how rigorous the clinical testing will have to be to establish safety and efficacy — the hallmarks of a PMA decision.
There is absolutely no other way.
And, even if developers open channels of communication with the FDA, there is no assurance those channels will be productive.
Bottom line — despite the extraordinary promise of artificial intelligence and the obvious value such products could bring, their routine use may be a long way off.
Editor’s note: This is the first blog in a series of four by industry consultant Greg Freiherr on Machine Learning and IT.
Related Content
News | Artificial Intelligence
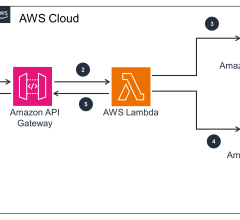
July 26, 2024 — GE HealthCare and Amazon Web Services, Inc. (AWS), an Amazon.com, Inc. company, announced a strategic ...
 July 26, 2024
July 26, 2024 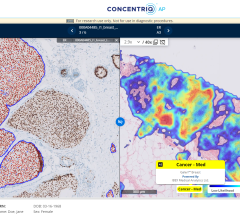
News | Digital Pathology
July 24, 2024 — Proscia, a developer of artificial intelligence (AI)-enabled digital pathology solutions for precision ...
July 24, 2024 
News | RSNA
July 23, 2024 — Professional registration is open for RSNA 2024, the world’s largest radiology forum. This year’s theme ...
July 23, 2024 
News | Artificial Intelligence
July 23, 2024 — Researchers at the National Institutes of Health (NIH) found that an artificial intelligence (AI) model ...
July 23, 2024 
News | Digital Pathology
July 12, 2024 — AGFA HealthCare, a global leader in healthcare imaging management solutions, announced that Enterprise ...
July 12, 2024 
News | Digital Pathology
July 12, 2024 — Diagnosing cancer and providing the personalized therapy it often requires, is a collaborative effort ...
July 12, 2024 
Feature | Imaging Technology News - ITN
Be sure to check out the latest digital edition of Imaging Technology News (ITN), featuring the Mobile C-arm Systems ...
July 11, 2024 
Feature | PACS | By Michael J. Cannavo
Back in 1966, Joni Mitchell sang these words in her song “Both Sides Now:” I’ve looked at clouds from both sides now ...
July 08, 2024 
Feature | Radiology Business
ITN conducts a bi-monthly survey to its readers on a variety of topics, which is used to create the Last Read, a unique ...
July 08, 2024 